<strike id="m6w4m"></strike>


地址:廣州市南沙區大崗鎮智新一路8號聯東U谷廣州南沙國際企業港7棟5樓
產品咨詢:13392693259
耗材電話:18998328368
售后服務:020-87684117
傳 真:020-87684846
Email:info@gdana.com
網址:http://www.alidw.com/
1 范圍
本標準規定了電子信息產品中含有的鉛(Pb)、汞(Hg)、鎘(Cd)、六價鉻[Cr(Ⅵ)]、多溴聯苯(PBB)和多溴二苯醚(PBDE)六種限用的有毒有害物質或元素的檢測方法。
本標準適用于《管理辦法》定義的電子信息產品。
2 規范性引用文件
下列文件中的條款通過本標準的引用而成為本標準的條款。凡是注日期的引用文件,其隨后所有的修改單(不包括勘誤的內容)或修訂版均不適用于本標準,然而,鼓勵根據本標準達成協議的各方研究是否可使用這些文件的最新版本。凡是不注日期的引用文件,其最新版本適用于本標準。
SJ/T11363 -2006 電子信息產品中有毒有害物質的限量要求。
3 術語和定義
本標準采用下列術語和定義。
3.1 電子信息產品
electronic information products EIP 采用電子信息技術制造的電子雷達產品、電子通信產品、廣播電視產品、計算機產品、家用電子產品、電子測量儀器產品、電子專用產品、電子元器件產品、電子應用產品以及電子材料產品等產品及其配件。
3.2 物質 substance
由自然界中存在的化學元素組成的單質或化合物。
3.3 篩選 screening
初步量化被檢物中待測元素濃度的一種分析方法。
3.4 聚合物材料 polymeric materials
通過鑄模或壓延能被制成薄膜或各種形狀的合成或半合成有機高分子濃縮材料。
注:聚合物材料包括聚乙烯,聚氯乙烯,環氧樹脂,聚酰胺,聚碳酸酯,ABS 樹脂,聚苯乙烯等。
3.5 金屬材料 metallic materials
金屬元素的單質或混合物,包括所有鋼鐵、有色金屬或合金材料。
注:金屬材料有鐵合金,鎳合金,錫合金,鋁合金,鎂合金,銅合金,鋅合金,貴金屬合金等。
3.6 專用電子材料 special electronic materials
在電子信息產品中所使用的一些特殊材料,由金屬材料、有機材料、無機非金屬材料中兩種或三種材料組成的混合物。
注:專用電子材料有線路板基材、導電膠、半導體功能材料等。
3.7 無機非金屬材料 inorganic nonmetallic materials
以某些元素的氧化物、碳化物、氮化物、硼化物以及硅酸鹽、鋁酸鹽、磷酸鹽、硼酸鹽等物質組成的材料。是除有機高分子材料和金屬材料以外的所有材料的統稱。
注:無機非金屬材料有玻璃、陶瓷等材料。
3.8 基體 matrix
含有或附有被分析物的材料或物質。
3.9 均勻材料 homogeneous materials
由一種或多種物質組成的各部分均勻一致的材料。
3.10 檢測單元 test unit
可以直接提交進行精確檢測的不需要進一步機械拆分的樣品。
3.11 X射線熒光光譜法 X-Ray fluorescence spectrumetry
XRF 用一束X射線或低能光線照射待測試樣,使之發射特征X射線而對物質成分進行定性和定量分析的方法。按激發、色散和探測方法的不同,分為波長散射-X射線熒光光譜法和能量散射-X射線熒光光譜法。
3.12 波長散射-X射線熒光光譜法 wavelength dispersive X-ray fluorescence spectru metry WD-XRF
試樣中被測元素的原子受到高能輻射激發而引起內層電子的躍遷,同時發射出具有一定特征波長的X射線,根據測得譜線的波長和強度來對被測元素進行定性和定量分析。
3.13 能量散射-X射線熒光光譜法 energy dispersive X-ray fluorescence spectrumetr y ED-XRF
試樣中被測元素的原子受到高能輻射激發而引起內層電子的躍遷,同時發射出具有一定能量的X射線,利用具有一定能量分辨率的X射線探測器探測試樣中被測元素所發出的各種能量特征X射線,根據探測器輸出信號的能量大小和強度來對被測元素進行定性和定量分析。
3.14 背景 background
疊加在分析線上的連續譜,主要來自試料對入射輻射的散射。
3.15 分析線 analyt e lines
需要對其強度進行測量并據此判定被分析元素含量的特征譜線。
3.16 檢出限 limit of detection
在一定置信水平下能檢出的最低含量。
3.17 干擾線 interference lines
與分析線重疊或部分重疊,從而影響對分析線強度進行準確測量的譜線。
3.18 基體效應 matrix effects
試料的化學組成和物理-化學狀態對分析線強度的影響,主要表現為吸收-增強效應、顆粒度效應、表面光潔度效應、化學狀態效應等
。
3.19 氣相色譜-質譜聯用法 gas chromatography - mass spectrometry GC-MS
將氣相色譜儀與質譜儀連接起來,利用氣相色譜高效的分離能力與質譜的特征檢測來對有機化合物進行定性與定量分析的方法。
3.20 電感耦合等離子體原子發射光譜法 inductively coupled plasma atomic emission spe ctrometry ICP-AES/OES
利用高頻等離子體使試樣原子化或者離子化,通過測量激發原子或離子的能量對應的波長來確定試 樣中存在的元素。
3.21 電感耦合等離子體質譜法 inductively coupled plasma mass spectrometry ICP-MS
通過高頻等離子體使試樣離子化的方法確定試樣所含的目標元素。用質譜儀測出產生的離子數量,并由目標元素的質/荷比來分析目標元素及其同位素。
3.22 原子吸收光譜法 atomic absorption spectrometry AAS
用火焰或化學反應等方式將欲分析試樣中待測元素轉變為自由原子,通過測量蒸氣相中該元素的基態原子對特征電磁輻射的吸收,以確定化學元素含量的方法。
3.23 冷蒸氣原子吸收光譜法 cold vapour generation atomic absorption spectromet ry CVAAS
將欲分析試樣中的汞離子,還原成自由原子,通過測量該蒸氣相中的基態原子對特征電磁輻射的吸收,以確定汞元素含量的方法。
3.24 原子熒光光譜法 atomic fluorescence spectrometry AFS
利用原子熒光譜線的波長和強度進行物質的定性與定量分析的方法。
4 檢測方法概述
4.1 檢測方法的內容
各種有毒有害物質或元素的檢測方法由如下6個部分組成:
——范圍;
——方法概要;
——儀器設備;
——試劑;
——制樣方法;
——測試步驟。
4.2 檢測方法流程 標準的檢測方法流程見圖1。其中,檢測單元為SJ/T××××-200×中規定的EIP-A/B/C。

4.3 篩選檢測方法
用能量散射X射線熒光光譜法(ED-XRF)或波長散射X射線熒光光譜法(WD-XRF)對試樣中目標物進行測試,可以是直接測量樣品(不破壞樣品),也可以是破壞樣品使其達到“均勻材料”(機械破壞制樣)后測試。很多有代表性的均勻材料(如塑料,合金,玻璃等)樣品可以不破壞樣品直接進行篩選,而由多種材料組成的復雜樣品(如組裝印制電路板)則須進行機械破壞性樣品制備。機械破壞性樣品制備既適用于篩選也適用于精確檢測方法。機械破壞性制樣的程序步驟見附錄A。
注意:第5 章詳細介紹的用XRF光譜法進行篩選的方法盡管是一種快速且節省資源的分析手段,但這種分析技術在獲得結果的適用性和應用方面存在一定的局限性。
篩選分析可以把樣品分成三個基本類別:
—— 合格(P):試樣中目標物的濃度低于允許值。
—— 不合格(F):試樣中目標物的濃度高于允許值。
—— 不確定(X):試樣中目標物的濃度在允許值附近,因為沒有肯定的合格與否的結果而需要進一步檢測。
通過篩選后,合格的樣品即是符合要求;不合格的樣品則不符合要求;而對于無定論的樣品則需用
4.4規定的精確測試程序來驗證該樣品是否符合要求。
4.4 精確測試方法
精確測試方法是運用多種分析方法來分析有機材料、金屬材料、無機非金屬材料以及專用電子材料中受限物質的濃度。表1是精確測試方法的簡單概要,詳細內容分別見第6章到第8章。
表1 精確測試方法簡單概要

5 用X射線熒光光譜儀對電子信息產品中有毒有害物質進行篩選的測試方法——此次省略具體參考原文。
6 電子信息產品中多溴聯苯(PBB)和多溴二苯醚(PBDE)的測試方法
6.1 范圍
本測試方法適用于在電子信息產品中所使用的聚合物材料和電子專用材料中多溴聯苯(PBB)和多溴二苯醚(PBDE)的測試,測試濃度范圍為(100~20000)mg/kg。如果采取適當的濃縮和凈化步驟,本方法可測試更低濃度的樣品。
6.2 方法概要
本方法采用索氏提取法從聚合物材料和電子專用材料中提取多溴聯苯和多溴二苯醚,通過適當的稀釋后,使用氣相色譜-質譜法(GC-MS)選擇離子監測模式(SIM)來測試提取液中多溴聯苯和多溴二苯醚,然后計算出它們在聚合物材料中的含量。
6.3 儀器設備
6.3.1 設備
a) 實驗用通風櫥;
b) 電子分析天平,精確到0.1mg;
c) 玻璃器皿;
d) 帶冷凝器的索氏提取裝置;
e) 用來加熱索氏提取裝置中用的燒瓶的加熱裝置;
f) 粉碎裝置:包括切割機或剪刀、研磨機、冷凍粉碎設施等裝置,主要用于樣品的粉碎;
g) 加熱爐 (可加熱到400℃以上);
h) 烘箱(可持續升溫至105℃~250℃);
i) 18目標準篩(篩孔直徑1mm)。
6.3.2 儀器
a) 氣相色譜儀:帶有程序升溫控制系統;
b) 毛細管柱:(10~30)m(長)×0.25mm(內徑)×0.1μm(膜厚)的熔融石英毛細管柱(DB-5或其他經證明同樣適用的色譜柱);
c) 質譜儀:能產生能量為70eV的電子用于電離被分析物,應能掃描(50~1000)m/z的區域;
d) 數據分析系統:能采集、記錄、儲存以及處理MS數據。
6.4 試劑
a) 溶劑:丙酮、甲苯、環己烷、正己烷、甲醇、二氯甲烷、異辛烷、壬烷,分析純;
b) 標準儲備液:多溴聯苯(PBB)和多溴二苯醚(PBDE)同分異構體的混合標準溶液;
c) 校準標準溶液:用標準貯備液至少需配制5種不同濃度的校準標準溶液,其中最低濃度應稍高于本測試方法的檢測限;
d) 內標溶液:對于多溴二苯醚(PBDE)的分析,用13 C12 標記的BDEs或 4,4’- 二溴八氟聯苯(f)或十氘代菲(Phenathrene D10)或十氯聯苯作為內標物質;對于多溴聯苯(PBB)的分析,可把 13 C12 標記的多溴聯苯(PBB)或4,4’-二溴八氟聯苯(f)或十氘代菲(Phenathrene D10)或十氯聯苯 作為內標物質;
e) 液氮:工業級。
6.5 樣品制備
6.5.1 樣品的粉碎按附錄A將電子信息產品拆解成為各種材料樣品,用剪刀或切割機(或其他方式)將樣品制成小于10mm×10mm×10mm小塊,液氮冷凍后用粉碎機(或采用其他等同效果的粉碎方法)將樣品粉碎成粒徑小于1mm的顆粒(過18#標準篩),混合均勻。
6.5.2 樣品液的提取
稱取上述混勻的樣品0.1g~0.2g,精確到0.001g,放入纖維素套筒中,置于索氏提取裝置中,加入50mL~200mL提取甲苯或其他溶劑[6.4a)],同時加入內標溶液[6.4d)],然后加入1~2粒沸石,安裝好索氏提取裝置,加熱提取(4~24)h,每小時至少6個循環。待溶液冷卻后,定容到適當體積的容量瓶中。該溶液可以直接用于測試步驟。如果試樣中待測物的濃度超過校正曲線的范圍,適當稀釋后再測試。
6.6 測試步驟
6.6.1 氣相色譜分析條件a)
a) 進樣口溫度:(250~320)℃;
b) 柱溫箱起始溫度及保持時間:100℃,保持(1~3)min;
c) 柱溫程序升溫條件:由(100~320)℃以(5~20)℃/min程序升溫,最后恒溫5min;
d) 載氣:氦氣,流量:(1~2)mL/min;
e) 氣相色譜-質譜接口溫度:320℃;
f) 進樣方式:(脈沖)不分流進樣;
g) 進樣量:(1~2)μL。
6.6.2 質譜分析條件
a) 電離方式:EI;
b) 電子能量:70eV;
c) 離子源溫度:(250~300)℃;
d) 分辨率:大于800(最好大于1000);
e) 分析模式:選擇離子監測(SIM),監測的離子分別見表3和表4。
6.6.3 樣品分析
a) 啟動儀器,待儀器穩定;
b) 根據儀器要求,用儀器校準樣品對儀器進行校準;
c) 建立標準曲線:依次將不同濃度的校準溶液(6.4 c)注入氣相色譜-質譜儀中,以校準溶液濃度為橫坐標,以校準樣的色譜峰面積為縱坐標建立標準曲線;
d) 樣品測試:按建立標準曲線時注入的體積將樣品提取液注入氣相色譜-質譜儀中,根據表3和表4給定的離子峰來確定試樣中含有的PBB和PBDE。然后根據峰面積,從標準曲線上計算出相對應的PBB和PBDE的含量。如果不需要計入十溴二苯醚,則可在統計或計算時去除。
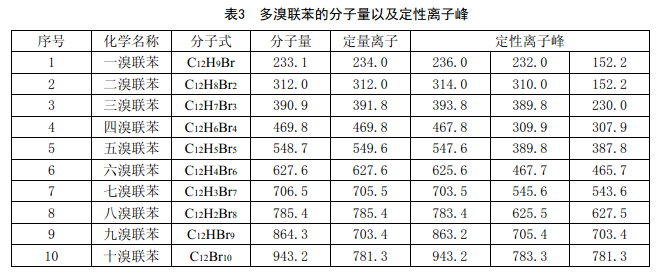
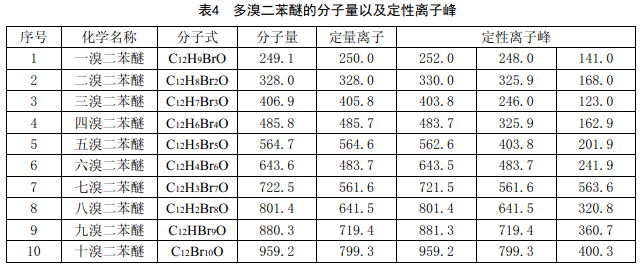
6.6.4 結果計算
試樣中多溴聯苯(PBB)和多溴二苯醚(PBDE)的結果計算按下式:
和多溴二苯醚(PBDE)")
6.6.5 精密度 在重復性條件下獲得的兩次獨立測試結果的絕對差值不得超過算術平均值的20%。
7 電子信息產品中鉛(Pb)、鎘(Cd)以及汞(Hg)的測試方法
7.1 電子信息產品中鉛(Pb)、鎘(Cd)的測試方法
7.1.1 范圍
本方法適用于電子信息產品中所使用的聚合物材料、金屬材料、電子專用材料以及無機非金屬材料中鉛和鎘含量的測試。
7.1.2 方法概要
稱取適量樣品,以微波消解、酸消解和灰化等方法處理后制成均勻液體。利用電感耦合等離子體原子發射光譜法(ICP-AES/OES)、電感耦合等離子體質譜法(ICP-MS)或原子吸收光譜法(AAS)測試樣品溶液中的鉛(Pb)和鎘(Cd)的濃度。
7.1.3 儀器設備
a) 原子吸收光譜儀(AAS);
b) 電感耦合等離子體原子發射光譜儀(ICP-AES/OES);
c) 電感耦合等離子體質譜儀(ICP-MS);
d) 實驗室用各種玻璃器皿;
e) 加熱裝置;
f) 馬弗爐;
g) 微波消解系統,配有高壓消解罐;
h) 電子分析天平,精確到0.1mg;
i) 坩鍋,50mL或150mL;
j) 耐氫氟酸容器;
k) 本生噴燈或相似的煤氣噴燈。
7.1.4 試劑
除非另有說明,在分析中僅使用認可的優級純以上試劑和 18M Ω 去離子水或相當純度的去離子水。
a ) 硝酸:ρ約1.4g/mL, 65%;
b) 鹽酸: ρ約1.16g/mL, 37%;
c) 過氧化氫:ρ約1.10g/mL,30%;
d) 硫酸: ρ約1.84g/mL,95%;
e) 氫氟酸:濃度大于40%;
f) 氫溴酸:ρ約1.48g/mL,47%~49%;
g) 高氯酸:ρ約1.67g/mL,70%;
h) 磷酸: ρ約1.69g/mL,大于85%;
i) 硼酸
j) 混合酸1(3份鹽酸+1份硝酸);
k) 混合酸2(3份鹽酸+1份過氧化氫);
l) 混合酸3(1份硝酸+3份氫氟酸);
m) 混合酸4(2份氫氟酸+1份硝酸+2份水);
n) 鉛標準溶液,濃度為1000μg/mL;
o) 鎘標準溶液,濃度為1000μ g/mL;
p) 鈧標準溶液,濃度為1000μg/mL;
q) 釔標準溶液,濃度為1000μg/mL;
r) 銠標準溶液,濃度為1000μg/mL。
7.1.5 樣品制備
7.1.5.1 樣品的粉碎
按附錄A將電子信息產品拆解成為各種材料樣品,將樣品制成小于10mm×10mm×10mm小塊。對金屬材料和無機非金屬材料可直接進行下一步工作,而對聚合物材料和電子專用材料,需繼續粉碎成粒徑小 于1mm的顆粒狀或粉末狀固體樣品,然后混合均勻備用。
7.1.5.2 金屬材料樣品的制備
7.1.5.2.1 酸消解法
稱取(0.1~0.5)g(可根據被檢元素的含量調整稱樣量)樣品于燒杯中,精確至0.0001g。根據金屬材料基體的不同,加入適量7.1.4中指定的酸(或適當比例的混合酸),加熱至試樣完全溶解。轉移至 50mL容量瓶,用水定容至刻度以備分析。
推薦采用鹽酸、硝酸或其混合酸溶液[7.1.4 l),m)]。當試樣難以消解時,需采用氫氟酸、高氯酸或硫酸等進一步溶解。在用氫氟酸時,需使用PTFE/PFA燒杯和其他耐氫氟酸的裝置。
注意:在溶解過程中,可能會產生沉淀(硫酸鉛,硫酸鋇,氯化銀,三氧化二鋁或氫氧化鋁等)因此需確定所選用的溶解方法不會造成目標元素的損失。若殘留物中含有目標分析物,需采用其他方法進行試樣溶解(如堿熔法或密封壓力容器法)。
a)一般合金方法
稱取(0.1~0.5)g(可根據被檢元素的含量調整稱樣量)樣品于燒杯中,精確至0.0001g。緩緩加入10mL鹽酸與硝酸的混合酸1(可根據基體成份進行適當調整),低溫加熱使之溶解完全。冷卻至室溫,轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦(Ti),鉭(Ta),鈮(Nb),鎢(W)時(該信息可從第5章的篩選試驗中獲得),需再加入1mL氫氟酸以保證試樣消解完全。
b)錫合金方法
稱取(0.1~0.5)g(可根據被檢元素的含量調整稱樣量)樣品于燒杯中,精確至0.0001g。緩緩加入10mL鹽酸與硝酸的混合酸1(可根據基體成份進行適當調整),蓋上表面皿,待劇烈反應后,微熱至試樣完全溶解。稍冷,移去表皿,沿杯壁加入10mL硫酸,加熱蒸發至剛冒SO3白煙,冷卻。
沿杯壁加入20mL氫溴酸,充分混勻,加熱至剛冒SO3白煙。重復3次。冷卻至室溫,加入10mL硝酸溶解鹽類。冷卻,轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法, 稀釋成相應濃度的樣品溶液。
或者,稱取0.5g樣品,精確至0.0001g,加入10mL鹽酸與過氧化氫的混合酸2(可根據基體成份進行適當調整)溶解試樣。
當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦(Ti),鉭(Ta),鈮(Nb),鎢(W)時(該信息可從第5章的篩選試驗中獲得),需再加入1mL氫氟酸以保證試樣消解完全。
7.1.5.2.2 微波消解法
稱取0.10g樣品,精確至0.0001g,置于消解罐中,加入5mL適宜比例的鹽酸與硝酸的混合酸1(可根據基體成份進行適當調整)。當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦(Ti),鉭(Ta),鈮(Nb), 鎢(W)時(該信息可從第5章的篩選試驗中獲得),再加1mL濃氫氟酸。
待試樣反應一段時間后,將整個消解罐置于微波消解儀中,按照儀器操作方法對樣品進行微波消解,直至樣品被完全溶解。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
7.1.5.3 無機非金屬樣品的制備
通常無機非金屬樣品中含有硅等非金屬元素,在樣品制備過程中需加入強腐蝕性的氫氟酸,應采用耐氫氟酸器皿。
稱取0.10g樣品,精確至0.0001g,置于消解罐中。加入3mL適宜比例的鹽酸與硝酸的混合酸1和3mL氫氟酸。待試樣反應一段時間后,將整個消解罐放入微波消解儀中,按照儀器操作方法對試樣進行微波消解,直至試樣被完全溶解。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
7.1.5.4 聚合材料樣品的制備
7.1.5.4.1 灰化法
a) 當樣品中不含鹵素化合物時(該信息可從第5章的篩選試驗中獲得):
稱取(0.1~0.5)g(可根據基體成份進行適當調整)經粉碎后的樣品于坩堝中,精確至0.0001g。
坩堝置于耐熱絕熱板的孔中,緩慢加熱至焦化狀態,逐漸加溫直至揮發的消解產物被充分排出,僅剩余最后只留下碳化物殘渣。將坩堝移入(450±25)℃的馬弗爐中,爐門輕微開啟以提供足夠的空氣來氧化碳,直至剩余干凈的灰分。將坩堝及其里面的物質從爐中取出,冷卻至室溫。加入5mL硝酸,溶解,轉移至50mL容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
b) 當樣品中含有鹵素化合物時(該信息可從第5章的篩選試驗中獲得):
稱取(0.1~0.5)g(可根據基體成份進行適當調整)經粉碎后的樣品于坩堝中,精確至0.0001g。
加入(10~15)mL的硫酸,將坩堝置于電熱板或砂浴上緩慢加熱,直到塑料熔化并變黑。再加入5mL硝酸,繼續加熱至塑料完全降解并產生白煙。冷卻,將坩堝移入(450±25)℃的馬弗爐中, 試樣被蒸發、干燥和灰化,直至碳被完全燃燒。將坩堝及其里面的物質從爐中取出,冷卻至室 溫。加入5mL硝酸,溶解,轉移至50mL容量瓶中,用水定容至刻度以備分析。根據所采用的分析 方法,稀釋成相應濃度的樣品溶液。
7.1.5.4.2 酸消解法
本方法只適用于鎘的測試。
注意:因在使用硫酸時會生成硫酸鉛沉淀而導致鉛的損失,所以本方法不適用于鉛的測試。
a) 一般溶解方法
稱取(0.1~0.5)g(可根據基體成份進行適當調整)經粉碎后的樣品于燒瓶中,精確至0.0001g。
加入5mL硫酸和1mL硝酸,加熱燒瓶至試樣灰化并產生白煙。停止加熱,加入少量硝酸(約0.5mL),繼續加熱至產生白煙。重復以上加熱過程,直至消解液變為淺黃色。
冷卻10min后,分若干次加入過氧化氫,總量不超過10mL,再次加熱至產生白煙。冷卻,轉移至50mL容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
b)當樣品消解不完全或樣品中含有二氧化硅、鈦等元素時(該信息可從第5章的篩選試驗中獲得):
稱取(0.1~0.5)g(可根據基體成份進行適當調整)經粉碎后的樣品于燒瓶中,精確至0.0001g。
加入5mL硫酸和1mL硝酸,加熱燒瓶至試樣灰化并產生白煙。停止加熱,加入少量硝酸(約0.5mL),繼續加熱至產生白煙。重復以上加熱過程,直至消解液變為淺黃色。
冷卻10min后,分若干次加入過氧化氫,總量不超過10mL,再次加熱至產生白煙。冷卻,將溶液移入氟碳樹脂罐中。加入5mL氫氟酸,并加熱直至產生白煙。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
7.1.5.4.3 微波消解法
稱取0.10g經粉碎后的樣品,精確至0.0001g,置于消解罐中,加入5mL硝酸、1mL過氧化氫。當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦(Ti),鉭(Ta),鈮(Nb),鎢(W)時(該信息可從第5章的篩選試驗中獲得),再加入1mL氫氟酸。將整個消解罐置于微波消解儀中,按照儀器操作方法對樣品進行微波消解,直至試樣被完全溶解。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
注意:過氧化氫可與易氧化材料發生快速而猛烈的反應。當樣品中可能含有大量易氧化有機成分時,不宜添加過氧化氫。
7.1.5.5 電子專用材料樣品的制備
稱取0.10g經粉碎后的樣品,精確到0.0001g,置于消解罐中,加入5mL適宜比例的鹽酸與硝酸的混合酸1(可根據基體成份進行適當調整)。當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦(Ti),鉭(Ta),鈮(Nb),鎢(W)時(該信息可從第5章的篩選試驗中獲得),再加入1mL氫氟酸。待試樣反應一段時間后,將整個消解罐置于微波消解儀中,按照儀器操作方法對樣品進行微波消解,直至試樣被完全溶解。
將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應濃度的樣品溶液。
7.1.6 測試步驟
7.1.6.1 空白溶液的制備
隨同7.1.5中樣品的制備一起全程制備空白溶液。
7.1.6.2 鉛和鎘校準溶液的配制
因各種分析儀器存在不同程度的基體效應問題,在配制校準溶液時應根據待測材料的種類分別采用外標法(基體匹配)、內標法或標準加入法配制校準溶液。
制備空白校準溶液與至少三種濃度的校準溶液。
當采用內標法時,可在配制時向溶液中加入內標元素,或在測試過程中采用儀器在線加入內標。對于ICP-AES/OES法,可選取鈧(Sc)或釔(Y)元素作內標;對于ICP-MS法,可選取銠(Rh)元素作內標。內 標元素的濃度與待測元素的濃度相當。
7.1.6.3 鉛和鎘校準曲線的繪制
a) ICP-AES/OES方法按濃度由低到高的順序測量校準系列溶液中目標元素的光譜發射強度讀數。根據需要測試內標元素的發射強度讀數。
采用外標法時,以校準溶液的濃度為橫坐標,以信號強度為縱坐標繪制校準曲線。
采用內標法時,以校準溶液的濃度為橫坐標,以待測元素的信號強度與內標元素的信號強度比為縱坐標繪制校準曲線。
分析時所選擇的譜線及可能存在的干擾:見表5。


當存在共存物質干擾時,應選取不會干擾測試的任一波長,或采用適當的方法消除干擾。
b) ICP-MS方法
按濃度由低到高的順序測量校準系列溶液中目標元素同位素的計數。根據需要測試內標元素同位素的計數。
采用外標法時,以校準溶液的濃度為橫坐標,以信號強度為縱坐標繪制校準曲線。
采用內標法時,以校準溶液的濃度為橫坐標,以待測元素的信號強度與內標元素的信號強度比為縱坐標繪制校準曲線。
分析時所選擇的同位素及可能存在的干擾見表6。

當存在共存物質干擾時,應選取不會干擾測試的任一同位素,或采用適當的方法消除干擾。
c) AAS方法
按濃度由低到高的順序測量校準系列溶液中目標元素的光譜吸收強度讀數。以校準溶液的濃度為橫坐標,以吸收強度為縱坐標繪制校準曲線。
分析線:鎘(Cd) 228.8nm;鉛(Pb) 217.0nm或283.3nm。
吸光度讀數范圍:為了減少光度測量的誤差,吸光度讀數一般選在0.1~0.6之間,必要時可調節溶液的濃度或光程長度或擴展量程。當存在共存物質干擾時,應選取不會干擾測試的任一波長,或采用適當的方法消除干擾。
7.1.6.4 樣品分析
校準曲線建立后,測試空白溶液、樣品溶液。依據每個試樣的信號讀數,由校準曲線查得所對應的濃度。每個樣品應平行分析兩次,且兩次結果的相對標準偏差不應該高于10%。同時在每一批樣品中至少取一個樣品進行加標回收率的試驗,回收率應該在90%~110%之間。
對于AAS法而言,若樣品溶液的濃度高于校準溶液的最高點,應將樣品溶液進一步稀釋,使其濃度落于校準溶液的濃度范圍內。
將標準物質或校準溶液作為質控樣品,每10個樣品測試一次,計算測量的準確度。如若需要,應重新繪制校準曲線。
7.1.6.5 結果計算
被測元素含量以質量分數WM計,數值以%表示,按下式計算:


7.1.6.6 精密度
在重復性條件下獲得的兩次獨立測試結果的絕對差值不得超過算術平均值的10%。
7.2 電子信息產品中汞(Hg)的測試方法
7.2.1 范圍
本方法適用于在電子信息產品中所使用的聚合物材料、金屬材料、電子專用材料以及無機非金屬材料中汞含量測試。
7.2.2 方法摘要
稱取適量樣品,以微波消解、酸消解的方法處理后制成均勻樣品溶液。樣品溶液應在4℃保存以減少汞的揮發。為了長期貯存汞溶液,建議采用5.0%硝酸+0.05%重鉻酸鉀的介質。
利用冷原子吸收法(CVAAS)、原子熒光法(AFS)、電感耦合等離子體原子發射光譜法(ICP-AES)、電感耦合等離子體質譜法(ICP-MS)或原子吸收光譜法(AAS)測試樣品溶液中的汞濃度。采用CVAAS與AFS時,在分析之前汞(Hg)被還原成原子狀態。
7.2.3 儀器設備
a) 冷蒸氣原子吸收光譜儀 (CVAAS);
b) 電感耦合等離子體原子發射光譜儀(ICP-AES/OES);
c) 電感耦合等離子體質譜儀(ICP-MS);
d) 原子熒光光譜儀(AFS)
e) 加熱回流裝置:配有反應瓶、回流冷凝裝置和吸收裝置;
f) 實驗室用各種玻璃器皿;
g) 耐氫氟酸的容器;
h) 加熱裝置;
i) 微波消解系統,配有高壓消解罐;
j) 電子分析天平,精確到0.1mg;
注意:因汞極易被污染,實驗中的每一個步驟都應極其小心。所有的取樣、儲存與操作裝置均應無汞(Hg)。所有的器皿在室溫下以50%的硝酸浸泡24h,再以18 MΩ去離子水徹底清洗。
7.2.4 試劑
除非另有說明,在分析中僅使用認可的優級純以上試劑和18MΩ去離子水或相當純度的去離子水。
a) 硝酸:ρ約1.40g/mL,65%;
b) 鹽酸:ρ約1.19g/mL,37%;
c) 過氧化氫: ρ約1.10g/mL,30%;
d) 氫氟酸:濃度大于40%;
e) 硫酸:ρ約1.84g/mL,95%;
f) 硼酸,分析純;
g) 汞標準溶液,濃度為1000μ g/mL;
h) 氯化鈉-鹽酸羥胺溶液:每100mL水溶解12g氯化鈉及12g鹽酸羥胺;
i) 高錳酸鉀:5%水溶液,每100mL水溶解5g高錳酸鉀;
j) 氫氧化鈉,分析純;
k) 硼氫化鈉;
l) 1 %硼氫化鈉-氫氧化鈉0.05%溶液.:向1000mL的容量瓶中加入超純水,至接近刻度。再加0.5g氫氧化鈉,溶解后,加10.0g硼氫化鈉,攪拌、溶解,以水定容至刻度,現配現用;
m) BCR-680,BCR-681:塑料包裝和包裝材料中的認證參照材料;
7.2.5 樣品制備
7.2.5.1 樣品的粉碎
按附錄A將電子信息產品拆解成為各種材料樣品,用剪刀或切割機(或其他方式)將樣品制成小于10mm×10mm×10mm小塊。對金屬材料和無機非金屬材料可直接進行下一步工作,而對聚合物材料和電子 專用材料,需繼續粉碎成粒徑小于1mm的顆粒狀或粉末狀固體樣品,然后混合均勻以備下一步工作。
7.2.5.2 金屬材料樣品的制備
7.2.5.2.1 酸消解法
a) 樣品制備的一般方法
稱取約1g樣品于潔凈反應瓶中,精確至0.0001g,加30mL硝酸。反應瓶裝有回流冷凝裝置及含有 10mL 0.5mol/L硝酸溶液的吸收裝置。在室溫下消解1h,升溫至90℃,恒溫消解2h。冷卻至室溫, 將吸收管內的溶液合并于消解液中。轉移至250mL容量瓶,以5%的硝酸溶液定容至刻度制備成樣品溶液。
b) 含有鋯(Zr)、鉿(Hf)、鈦(Ti)、銅(Cu)、銀(Ag)、鉭(Ta)、鈮(Nb)或鎢(W)的材料的溶解
稱取約1g樣品于潔凈反應瓶中,加入20mL濃鹽酸和10mL濃硝酸。反應瓶裝有回流冷凝裝置及含 有10mL 0.5mol/L硝酸溶液的吸收裝置。在室溫下消解1h。升溫至(95±5)℃,消解15min。取下, 冷卻。
若試樣未被完全消解,反復加王水并加熱,直至試樣消解完全。每次加酸時,都應沿器壁加入,以將粘在壁上的試樣重新沖洗至溶液中。
待試樣完全消解后,向反應瓶中加入20mL水與15mL高錳酸鉀溶液。充分混勻,并在(95±5)℃加熱回流30min。冷卻到室溫,過濾,轉移至100mL容量瓶中。用水反復清洗反應瓶、冷凝器與 吸收裝置。將清洗液并入容量瓶中。可加入6mL氯化鈉-鹽酸羥胺溶液還原多余的高錳酸鉀4。用水稀釋至刻度,混勻制備成樣品溶液。
7.2.5.2.2 微波消解法
稱取0.10g樣品,精確至0.0001g,置于消解罐中,加入5mL適宜比例的鹽酸與硝酸的混合酸(鹽酸與硝酸比例通常為3+1,可根據基體成份進行適當調整)。當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦 (Ti),鉭(Ta),鈮(Nb),鎢(W)時(該信息可從第5章的篩選試驗中獲得),再加1mL濃氫氟酸。
待試樣反應一段時間后,將整個消解罐置于微波消解儀中,按照儀器操作方法對試樣進行微波消解,直至試樣被完全溶解。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應的濃度制備成樣品溶液。
7.2.5.3 無機非金屬樣品的制備
通常無機非金屬樣品中含有硅等非金屬元素,在樣品制備過程中需加入強腐蝕性的氫氟酸,因此要求采用耐氫氟酸器皿。稱取0.10g樣品,精確至0.0001g,置于消解罐中。加入3mL適宜比例的鹽酸與硝酸的混合酸(鹽酸與硝酸比例通常為3+1,可根據基體成份進行適當調整)、3mL濃氫氟酸。待試樣反應一段時間后,將整個消解罐放入微波消解儀中,按照儀器操作方法對試樣進行微波消解,直至試樣被完全溶解。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應的濃度制備成樣品溶液。
7.2.5.4 聚合材料樣品的制備
稱取0.10 g經粉碎后的樣品,精確至0.0001 g,置于消解罐中,加入5 mL硝酸、1mL過氧化氫。當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦(Ti),鉭(Ta),鈮(Nb),鎢(W)時(該信息可從第5章的篩選試驗中獲得),再加入1mL氫氟酸。將整個消解罐置于微波消解儀中,按照儀器操作方法對試樣進行微波消解,直至試樣被完全溶解。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應的濃度制備成樣品溶液。
注意:過氧化氫可與易氧化材料發生快速而猛烈的反應。當樣品中可能含有大量易氧化有機成分時,不宜添加過氧化氫。
7.2.5.5 電子專用材料樣品的制備
稱取0.10 g經粉碎后的樣品,精確到0.0001g,置于消解罐中,加入5 mL適宜比例的鹽酸與硝酸的混合酸(鹽酸與硝酸比例通常為3+1,可根據基體成份進行適當調整)。當樣品中含硅(Si),鋯(Zr),鉿(Hf),鈦(Ti),鉭(Ta),鈮(Nb),鎢(W)時(該信息可從第5章的篩選試驗中獲得),再加入1mL 氫氟酸。待試樣反應一段時間后,將整個消解罐置于微波消解儀中,按照儀器操作方法對試樣進行微波消解,直至試樣被完全溶解。將消解罐取出,冷卻至室溫,加入適量的硼酸絡合過量的氫氟酸(若采用耐氫氟酸的霧化器,可不加硼酸)。轉移至50mL的容量瓶中,用水定容至刻度以備分析。根據所采用的分析方法,稀釋成相應的濃度制備成樣品溶液。
7.2.6 測試步驟
7.1.6.1 空白溶液的制備
隨同7.2.5中樣品的制備一起全程制備空白溶液。
7.2.6.2 汞標準溶液的配制
汞標準溶液應貯存于惰性塑料容器中。濃度為1000μg/mL的汞溶液穩定期至多為一年。濃度小于1 μg/L的溶液應現用現配。
汞易被吸附在容器內壁,從而對標準溶液的穩定性造成較大影響。因此,建議加入幾滴5%的高錳酸鉀溶液以穩定該標準溶液。
因各種分析儀器存在不同程度的基體效應問題,在配制校準溶液時應根據待測材料的種類分別采用外標法(基體匹配)、內標法或標準加入法配制校準溶液。
制備空白校準溶液與至少三種濃度的校準溶液。
當采用內標法時,可在配制時向溶液中加入內標元素,或在測試過程中采用儀器在線加入內標。對于ICP-AES法,可選取鈧(Sc)或釔(Y)元素作內標;對于ICP-MS法,可選取銠元素(Rh)作內標。內標元素的濃度與待測元素的濃度相當。
7.2.6.3 汞校準曲線的繪制
a) ICP-AES/OES方法
按濃度由低到高的順序測量校準系列溶液中汞的光譜發射強度讀數。根據需要測試內標元素的發射強度讀數。
采用外標法時,以校準溶液的濃度為橫坐標,以信號強度為縱坐標繪制校準曲線。
采用內標法時,以校準溶液的濃度為橫坐標,以汞的信號強度與內標元素的信號強度比為縱坐標繪制校準曲線。
分析時所選擇的汞的譜線:194.227nm。
b) ICP-MS方法
按濃度由低到高的順序測量校準系列溶液中汞同位素的計數。根據需要測試內標元素同位素的計數。
采用外標法時,以校準溶液的濃度為橫坐標,以信號強度為縱坐標繪制校準曲線。
采用內標法時,以校準溶液的濃度為橫坐標,以汞的信號強度與內標元素的信號強度比為縱坐標繪制校準曲線。
內標法或標準加入法分析時所選擇汞的同位素:202m/z。
c) CVAAS方法
按濃度由低到高的順序測量校準系列溶液中汞的吸收強度讀數。以校準溶液的濃度為橫坐標,以汞的吸收強度為縱坐標繪制校準曲線。
吸光度讀數范圍:為了減少光度測量的誤差,吸光度讀數一般選在0.1~0.6之間,必要時可調節溶液的濃度或光程長度或擴展量程。
儀器參數:
光源:Hg無極放電燈或空心陰極燈;
波長:253.7nm;
光譜狹縫寬度:0.7nm;
吹掃氣:氮氣或者氬氣;
還原劑:1%硼氫化鈉-氫氧化鈉0.05%溶液(可根據需要適當調節濃度)。
d) AFS方法
按濃度由低到高的順序測量校準系列溶液中汞的熒光強度讀數。以校準溶液的濃度為橫坐標,以熒光強度為縱坐標繪制校準曲線。
熒光強度讀數范圍:為了減少測量的誤差,熒光強度讀數應落于儀器的線性范圍內,必要時可調節溶液的濃度。
儀器參數:
光源:Hg空心陰極燈;
電流:30mA;
波長:253.7nm;
負高壓:360V;
爐溫:800℃;
氬氣流量載氣:600mL/min,
屏蔽氣:1000mL/min;
還原劑:1%硼氫化鈉-氫氧化鈉0.05%溶液(可根據需要適當調節濃度)。
7.2.6.4 樣品分析
校準曲線建立后,測試空白溶液、樣品溶液。依據每個試樣的信號讀數,由校準曲線查得所對應的濃度。每個樣品應平行分析兩次,且兩次結果的相對標準偏差不應該高于20%。同時在每一批樣品中至少取一個樣品進行加標回收率的試驗,回收率應該在70%~130%之間。
對于CVAAS法與AFS法而言,若樣品溶液的濃度高于校準溶液的最高點,應將樣品進一步稀釋,使其濃度落于校準溶液的濃度范圍內。
當采用CVAAS法與AFS法時,應考慮到基體成分對氧化-還原反應的影響。
AFS法將標準物質或校準溶液作為質控樣品,每10個樣品測試一次,計算測量的準確度。如若需要,應重新繪制校準曲線。
7.2.6.5 結果計算
被測元素含量以質量分數WM計,數值以%表示,按下式計算:

7.2.6.6 汞污染防止
在使用各種方法或者器皿時須十分小心,從而將污染降至最低。以下預處理可在一定程度上避免樣品的污染:
a)僅采用蒸餾水或者去離子水。與水接觸的惰性塑料材料須小心處理。純水即使貯存于聚四氟乙烯容器(PTFE)中,也會在很短時間內從容器中浸出雜質;
b)制備樣品用的化學試劑是污染的主要來源,應使用不含汞的試劑;
c)樣品制備所需的化學試劑與還原劑在使用前,均需測量其空白值;
d)燒杯、滴管及容量瓶是金屬污染的主要來源,處理樣品時建議使用惰性塑料;
e)采用ICP-AES/OES、ICP-MS、AFS法時,當汞的濃度很高時,易產生記憶效應。因此在測試汞濃度較高的溶液時應盡量采用選擇記憶效應小的進樣系統,或將溶液稀釋。測量后應徹底清洗進樣系統。
7.2.6.7 精密度
在重復性條件下獲得的兩次獨立測試結果的絕對差值不得超過算術平均值的20%。
上一篇:GB/T22788-2008玩具表面涂層中總鉛含量的測定
下一篇:無

掃描二維碼
版權所有©廣州格丹納儀器有限公司 ![]() 粵公網安備 44011202000628號 粵ICP備14047970號
粵公網安備 44011202000628號 粵ICP備14047970號
分享到:







